清华大学・鲁志实验室科研项目及其转化寻求合作
目录:
- 清华大学・鲁志实验室科研项目及其转化寻求合作
- 项目一:针对癌症等复杂疾病的核酸检测项目 - AI驱动的cfRNA检测技术
- 项目二:RNA制药项目 - AI 驱动的RNA药物设计技术
- 相关专利
- 技术成果相关报道
- 联系方式
实验室简介
鲁志实验室(实验室主页: http://lulab.life.tsinghua.edu.cn)下属于清华大学生命科学学院,实验室成员包括博士生10余名和本科实习生若干,来自生物学、统计学、计算机科学等多学科。实验室依托于“生物信息学教育部重点实验室”和清华大学“合成与系统生物学研究中心”,拥有很好的人才支撑、硬件平台和合作科研环境。
我们实验室致力于发展生物信息学技术,并探索其在癌症、自身免疫等复杂疾病的精准诊疗上的具体应用。我们利用机器学习等人工智能技术,结合非编码RNA (ncRNA)为核心的多组学数据,来研究遗传信息是如何被编码在结构化的DNA和RNA分子之中,以及它们是如何在一个生命体系中相互作用、彼此调控。 “上工治未病”,我们的一个重要使命,是帮助人们更早期的发现和治疗疾病。我们相信,这种使命感以及为此付出的实践和努力,将帮助我们理解和治疗人类疾病,并最终认识和提高我们自己。
实验室负责人鲁志博士 (个人官方主页),清华大学生命科学学院长聘副教授(tenured)、特别研究员、博士生导师,国家基金委“优秀青年”基金、某青年人才项目、“霍英东”青年基金获得者。鲁志博士近20年来一直致力于非编码RNA相关的生物信息学研究,发表国际重要期刊文章~70篇 (Pulibcation List),包括通讯作者文章30余篇(ESI高被引论文4篇);文章所发表期刊包括 Science, Nature, Cell, PNAS, e-Life, Genome Biology等,总引用超过2万次。
实验室围绕着非编码RNA(ncRNA)主要有两个研究方向: 1)AI驱动的核酸模型;2)生信驱动的精准医疗。 详见 Projects 。
我们针对noncoding RNA的近20年的研究经验和主要成果如下图所示:

代表性在研项目
- 国家重点研发计划 重点专项,难治性自身免疫病精准诊治与创新疗法研究,2024.12-2027.11 课题组长
- 国家重点研发计划 重点专项,非编码RNA高通量解析技术与临床应用研发,2024.12-2027.11 课题骨干
- 国家自然科学基金 专项项目,82341101,基于AI技术的靶向RNA的药物设计,2024-2027 主持
- 国家自然科学基金 面上项目,82371855,自身免疫性疾病精准诊疗中基于非编码RNA组学和生物信息学的新方法研究,2024-2027 主持
- 国家自然科学基金 面上项目,3217040246,体液多组学数据整合的生物信息学研究及其在癌症无创检测上的应用,2022-2025 主持
- 国家自然科学基金 面上项目,81972798,针对癌症诊断和预后的新型体液exRNA标志物的研究,2020-2023 主持
- 国家“凤凰工程” 技术创新开放共享课题,2021-NCPSB-005,多组学整合的生物信息学研究,2021-2023 主持
- 清华大学精准医学研究院 精准医疗战略项目, 基于多组学数据的智能化中晚期肝癌转化治疗决策研究,2022-2025 主持
- 清华大学国强研究院 人工智能与机器人项目,2021GQG1020,生物调控网络知识启发下的新型AI模型的构建及其在癌症早诊上的应用,2022-2024 主持
- 清华大学春风基金,2021Z99CFY022,针对微生物感染的新型核酸检测技术研发,2021-2023 主持
- 合肥综合性国家科学中心大健康研究院 全球遴选项目, cfRNA为核心的多组学在肺癌筛查上的应用研究, 2024-2027 主持
- Bayer AI制药项目, AI driven drug design targeting RNAs,2023-2024 主持
项目一:针对癌症等复杂疾病的核酸检测项目 - AI驱动的cfRNA检测技术
基本信息
- 合作课题:针对癌症等复杂疾病的AI驱动的cfRNA检测技术
- 招募机构:
- 1. 医院、临检机构等
- 2. 基因检测公司等
- 3. 人工智能相关的实验室
- 4. 检测和测序技术相关的实验室
- 合作方式:
- 合作临床项目发现新靶标和新标志物
- 合作产业化项目开发和应用检测试剂盒
- 合作科研课题进行人工智能技术开发
- 合作科研课题进行检测和测序技术开发
- 发起组织:清华大学生命学院鲁志实验室 (Lab of RNA Bioinformatics)
- 有效时间:长期
研究目标和项目简介
- 研究目标:
该癌症检测项目的目的是要为癌症早期筛查、早期诊断以及预后治疗寻找到更好(精准、重复性高、经济上利于普及大众、操作上简单无创)的新型体液标志物(exRNA/cfRNA),并标准化其检测和解析流程,开发出新的液体活检技术。“上工治未病”,我们要帮助更多人更早地发现癌症。
- 项目简介:
本项目将基于基因组学和生物信息学,通过开发体液RNA微量测序技术和机器学习方法,在体液中发现和鉴定与癌症发生发展相关的新型exRNA标志物,应用于国内高致死癌症的早期诊断和预后辅助治疗。exRNA指的是胞外RNA,又称cfRNA,包括多种类型:miRNA,Y RNA,circRNA,lncRNA等。RNA标志物与DNA和蛋白标志物相比,具有更好的敏感性、组织特异性和多样性,为更好的临床检验带来了新的期望。我们在新型非编码RNA和生物信息学研究方面积累了丰富经验,基于此,我们将在癌症病人体液(如血液)中发现和分析标志癌症发生发展的新型exRNA/cfRNA,并整合现有标志物构建多重标志物的智能模型,在大样本上进行验证,建立具有更高精准度和可重复性的无创检验方法。
我们在非编码RNA(ncRNA)测序和生物信息学研究中积累了近20年的丰富经验,例如,我们在模式生物和肝癌样本中通过测序和生物信息学分析发现了很多新的lncRNA(Science 2010; Nature 2012; Nature 2014; Genome Biology 2017; Cell 2019; Cell Research 2020; PNAS 2020), 其中有不少具有很好的标志物特性。从2015年起,实验室开始大力发展针对体液无创检测技术的研究,我们已经克服了体液游离RNA易降解及微量建库的技术难题,开发了自主研发的超微量RNA测序技术i-SMART(专利号:201810607652X)、DETECTOR-seq (专利申请号:202210579444.X) (elife 2022, Cell Rep. Med. 2023, Clinic. & Tranl. Med. 2024),以及基于机器学习和人工智能的生物信息学方法RNAfinder(专利号:2016108069288)和exSEEK(专利申请号:202010618721.4)(Genome Res. 2011; Nucleic Acids Res. 2015; Nucleic Acids Res. 2017a),发现了一些新的exRNA标志物(专利号:2018110094643;专利号:202010927225.7) (Nature Communications 2017; Clinical Chemistry 2019; Theranositics 2021),积累了癌症相关的RNA数据库(著作权号:2016R11S367236)(Nucleic Acids Res. 2017b; Nucleic Acids Res. 2019;Nucleic Acids Res. 2022),为癌症无创检测试剂的开发提供了有力的支持。
合作方式
1) 合作发表科研文章
合作发现和研究体液中的新型exRNA/cfRNA,共同发表科研文章
- 方法学开发
- 共同开发针对多组学、多模态数据整合的人工智能模型
- 共同开发针对cfRNA(以及多组学)的高通量测序技术
- I期. 新型标志物的“发现和探索”阶段
- 贵单位构负责收集 少量(30-50例血样) 癌症病人和/或健康对照人群的血浆,妥善储存并运输;
- 我们负责进行预实验和分析,实验和分析费用由我们承担。
- II期. 新型标志物的“鉴定和验证”阶段
- 在前期预实验效果较好的前提下,还需要在 大量样本(200-500例血样) (最好部分血样能配套组织样本)中进行测序和验证;
- 实验和分析费用由双方共同承担,具体合作事宜面议。
样本收集与临床队列设计:
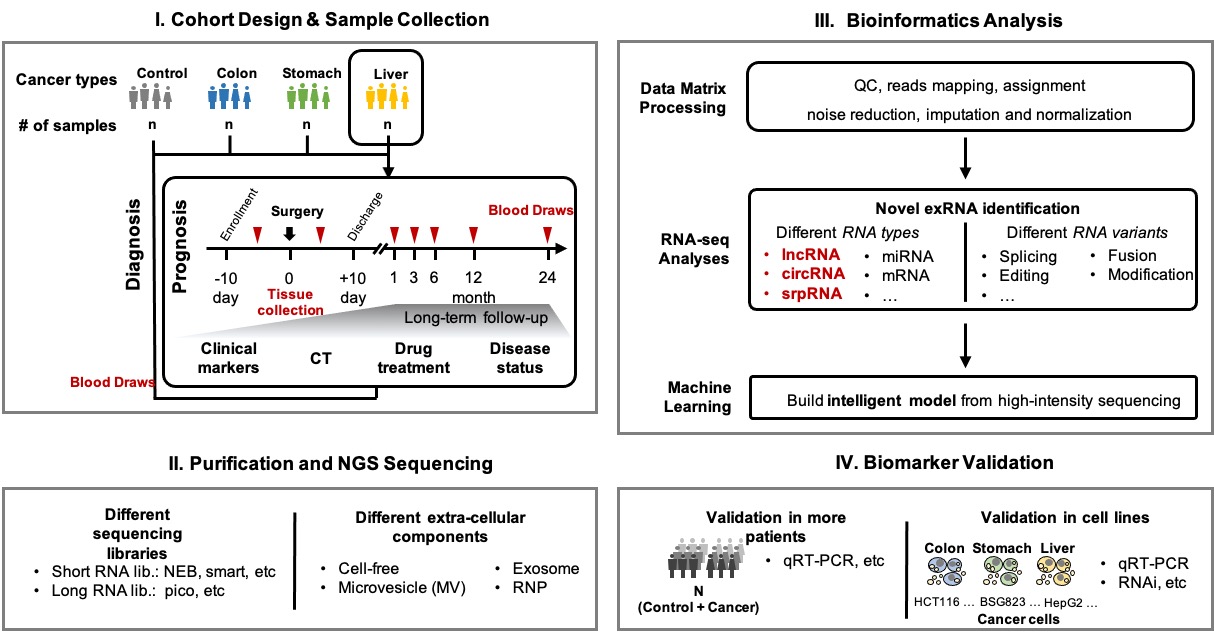
2) 合作开发试剂盒和申请器械证
我们提供相关试剂盒和检测方法的开发和生产方案
- III期. 新型标志物优化和试剂盒设计
- 我们负责从前期科研成果和自有的液体活检生物标志物数据库(注1)中根据贵单位需求筛选出证据等级和临床应用潜力较高的候选生物标志物,涵盖DNA,RNA,蛋白,代谢物等不同分子类型,我们将综合考虑标志物的一致性报道个数、已验证的样本数、生物学功能和诊断效果等;
- 贵单位负责对筛选的标志物在200-500例癌症病人和健康对照人群的血浆样本中进行二次验证;
- 我们负责对验证数据进行再分析,采用机器学习等人工智能方法优化出诊断模型,设计相应试剂盒;
- 贵单位进行相应试剂盒生产和推广。
- IV期. 大规模临床报证和产业化阶段
- 进行三类医疗器械证的注册、申报和推广,具体合作事宜面议,可以共同寻找其他投资和合作伙伴。
注1:
我们已经构建了一个包含 DNA,RNA,蛋白质和代谢物的整合性液体活检生物标志物数据库。该数据库包括:
- 获 FDA/CFDA 批准的近百个用于临床肿瘤诊断、指导用药和预后的生物标志物;
- 从 上千篇已发表科研文献中手动收集的具有临床应用潜能的生物标志物;
- 从公开发表的几千套液体活检高通量数据中鉴定出来的候选生物标志物。
我们的数据库涵盖了肺癌、乳腺癌、结直肠癌、肝癌、胰腺癌、胃癌、食管癌、脑胶质瘤、多发性骨髓瘤、前列腺癌、卵巢癌、肾癌、冠心病等31种人类疾病。同时,我们基于标志物的证据水平发展了一套标志物分级系统,给每一个标志物指定一个证据等级,帮助用户快速地认识所感兴趣的标志物的临床应用潜力。
3) 提供标志物发现和优化的实验方案
我们提供体液中新型标志物的发现、鉴定和优化方法 - 实验方案
我们在前期研究exRNA的过程中,参考最新的单细胞测序方法,摸索了一套可以从体液中高效率地捕获不同长度、不同类型的exRNA(又称为cfRNA)的实验流程。我们将根据贵单位需求开发相应试剂盒,可以帮助使用者从多种体液中进行exRNA/cfRNA的研究,涵盖了提取、纯化、捕获、扩增等一系列步骤的试剂及方法,可在1-2天内完成从体液获取到exRNA/cfRNA上机测序的全部准备,实用便捷。
包含small cfRNA/exRNA和total cfRNA/exRNA的全转录组捕获、富集和测序方案:
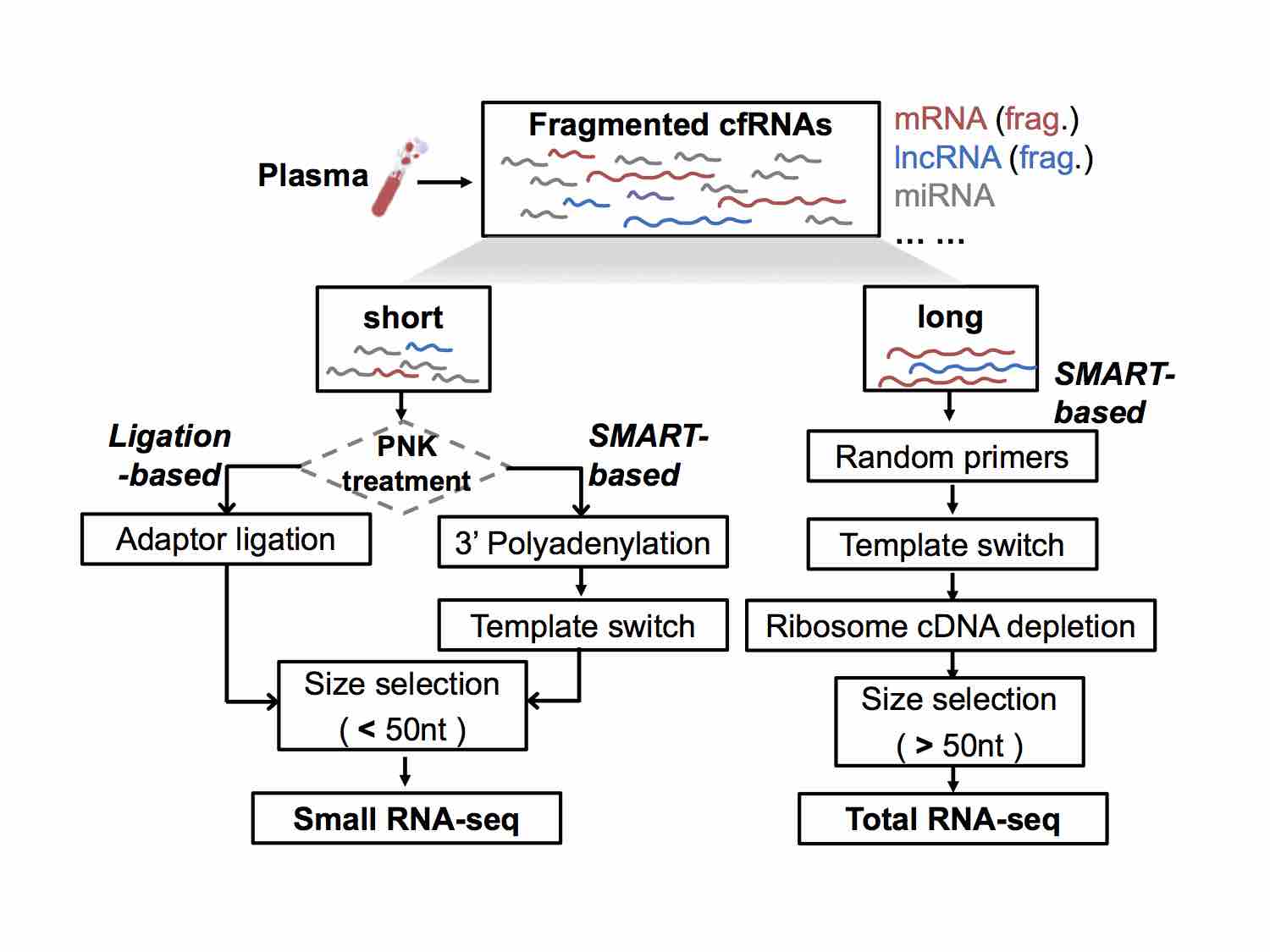
4) 提供标志物发现和优化的生信分析
我们提供体液中新型标志物的发现、鉴定和优化方法 - 生物信息学分析和软件定制。
我们在开展广泛应用于基因组学和癌症生物学的科学实践的同时,开发了一系列分析工具和平台。主要包括RNAfinder、RNAtarget、RNAstructurome、RNAmed四个系列:
- a) RNAfinder
- Ribowave: 用于核糖体测序数据(Ribo-seq)的分析,利用小波变换模型来提取核糖体3-nt周期(即信号频率)来使原始信号去噪声化,并精确定位其足迹信息 (Nucleic Acids Research 2018)。
- COME: 用于mRNA和lncRNA中蛋白编码潜能的分析 (Nucleic Acids Research 2017)。
- RNAfeature: 用于多个物种间非编码RNA保守性信息的分析 (Nucleic Acids Research 2015)。
- b) RNAtarget
- c) RNAstructurome
- RME/RNAex: 一种RNA二级结构预测算法,同时提供了多种类型的RNA二级结构实验数据 (Nucleic Acids Research 2015/2016)。
- d) RNAmed
合作方资质需求和已有合作单位
具有收集癌症病人和/或健康对照人群体液和/或组织样品的资质 ,或具有试剂盒开发及推广经验和资质的机构及公司。
- 已有合作单位:
- 北京 北京协和医院
- 北京 北京大学第一医院
- 北京 清华长庚医院
- 天津 天津肿瘤医院
- 上海 海军军医大学
- 成都 华西医院
- 。。。
项目二:RNA制药项目 - AI 驱动的RNA药物设计技术
基本信息
- 合作课题:AI驱动的RNA药物设计在疾病治疗上的应用
- 招募机构:
- 1. 制药公司、新药研发公司等
- 2. 人工智能相关的实验室
- 3. 药学和结构研究相关的实验室
- 4. CRO公司等
- 药物设计类型:
- 利用和优化我们的 AI 方法 (OligoFormer) 筛选和设计针对RNA序列的小RNA (siRNA) 药物
- 利用和优化我们的 AI 方法 (RNAsmol) 筛选和设计针对RNA结构的小分子药物
- 利用和优化我们的计算方法 (RBPgroup)和数据库 (POSTAR) 筛选和设计针对RNA结构的蛋白药物
- 合作方式:
- 科研课题:合作方提供人工智能技术,共同应用在RNA药物设计优化上
- 科研课题:合作方提供药学设计技术,共同应用在RNA药物设计优化上
- 科研课题:合作方针对我们预测出来的小分子或者小 RNA药物进行早期实验验证和优化
- 科研和产业化课题:合作方提供药筛平台进行药物筛选
- 产业化课题:合作方提供应用场景(例如针对特定疾病和组织提供递送siRNA的方法和手段)
- 发起组织:清华大学生命学院鲁志实验室(Lab of RNA Bioinformatics)
- 有效时间:长期
研究目标
我们在RNA-siRNA/shRNA结合,RNA-小分子结合,RNA-蛋白结合的计算设计上积累了大量科研经验,并应用在了HIV、HCV等病毒的靶标筛选上。希望能应用在癌症治疗、病毒感染治疗等疾病治疗上。
项目简介
我们和该项目相关的前期科研基础有(1第一作者,*最后通讯,*+:共同通讯):
- 1)RNA二级结构预测算法 (RNAstructure: Nucleic Acids Res. 20061,RNA 20091, Nucleic Acids Res. 2015*,2016* );
- 2)RNA-oligo/siRNA预测 (OligoWalk: Nucleic Acids Res. 2008a1,2008b1,2008c1; OligoFormer: Bioinformatics 2024b*)和shRNA 设计 (PNAS 2012);
- 3)RNA-蛋白结合预测 (POSTAR: Nuc. Acids. Res. 2017*, 2019*+, 2022*;RBPgroup: Genome Biology 2017*, Cell 2019 );
- 4)RNA-小分子结合预测
- 靶标病毒的小分子药物筛选(Nature Biotech. 2012);
- 小分子表型筛选计算方法(Brief. in Bioinformatics 2019*+);
- 靶标RNA的小分子药物AI设计(RNAsmol: preprint 2024*)。
Motivation: RNA interference (RNAi) has become a widely used experimental approach for post-transcriptional regulation and is increasingly showing its potential as future targeted drugs. However, the prediction of highly efficient siRNAs (small interfering RNAs) is still hindered by dataset biases, the inadequacy of prediction methods, and the presence of off-target effects. To overcome these limitations, we propose an accurate and robust prediction method, OligoFormer, for siRNA design. Results: OligoFormer comprises three different modules including thermodynamic calculation, RNA-FM module, and Oligo encoder. Oligo encoder is the core module based on the transformer encoder. Taking siRNA and mRNA sequences as input, OligoFormer can obtain thermodynamic parameters, RNA-FM embedding, and Oligo embedding through these three modules, respectively. We carefully benchmarked OligoFormer against six comparable methods on siRNA efficacy datasets. OligoFormer outperforms all the other methods, with an average improvement of 9% in AUC, 6.6% in PRC, 9.8% in F1 score, and 5.1% in PCC compared to the best method among them in our inter-dataset validation. We also provide a comprehensive pipeline with prediction of siRNA efficacy and off-target effects using PITA score and TargetScan score. The ablation study shows RNA-FM module and thermodynamic parameters improved the performance and accelerated convergence of OligoFormer. The saliency maps by gradient backpropagation and base preference maps show certain base preferences in initial and terminal region of siRNAs. Availability and implementation:The source code of OligoFormer is freely available on GitHub at: https://github.com/lulab/OligoFormer. Docker image of OligoFormer is freely available on the docker hub at https://hub.docker.com/r/yilanbai/oligoformer. - Bioinformatics 2024
RNA-targeting drug discovery is undergoing an unprecedented revolution. Despite recent advances in this field, developing data-driven deep learning models remains challenging due to the limited availability of validated RNA-small molecule interactions and the scarcity of known RNA structures. In this context, we introduce RNAsmol, a novel sequence-based deep learning framework that incorporates data perturbation with augmentation, graph-based molecular feature representation and attention-based feature fusion modules to predict RNA-small molecule interactions. RNAsmol employs perturbation strategies to balance the bias between true negative and unknown interaction space thereby elucidating the intrinsic binding patterns between RNA and small molecules. The resulting model demonstrates accurate predictions of the binding between RNA and small molecules, outperforming other methods with average improvements of ∼8% (AUROC) in 10-fold cross-validation, ∼16% (AUROC) in cold evaluation (on unseen datasets), and ∼30% (ranking score) in decoy evaluation. Moreover, we use case studies to validate molecular binding hotspots in the prediction of RNAsmol, proving the model’s interpretability. In particular, we demonstrate that RNAsmol, without requiring structural input, can generate reliable predictions and be adapted to many RNA-targeting drug design scenarios. - preprint 2024
RNA-binding proteins (RBPs) play key roles in post- transcriptional regulation. Accurate identification of RBP binding sites in multiple cell lines and tissue types from diverse species is a fundamental en- deavor towards understanding the regulatory mech- anisms of RBPs under both physiological and patho- logical conditions. Our POSTAR annotation pro- cesses make use of publicly available large-scale CLIP-seq datasets and external functional genomic annotations to generate a comprehensive map of RBP binding sites and their association with other regulatory events as well as functional variants. Here, we present POSTAR3, an updated database with im- provements in data collection, annotation infrastruc- ture, and analysis that support the annotation of post-transcriptional regulation in multiple species in- cluding: we made a comprehensive update on the CLIP-seq and Ribo-seq datasets which cover more biological conditions, technologies, and species; we added RNA secondary structure profiling for RBP binding sites; we provided miRNA-mediated degra- dation events validated by degradome-seq; we included RBP binding sites at circRNA junction re- gions; we expanded the annotation of RBP binding sites, particularly using updated genomic variants and mutations associated with diseases. POSTAR3 is freely available at http://postar.ncrnalab.org. - Nucleic Acids Res. 2022
The systematic identification of effective drug combinations has been hindered by the unavailability of methods that can explore the large combinatorial search space of drug interactions. Here we present multiplex screening for interacting compounds (MuSIC), which expedites the comprehensive assessment of pairwise compound interactions. We examined ~500,000 drug pairs from 1,000 US Food and Drug Administration (FDA)-approved or clinically tested drugs and identified drugs that synergize to inhibit HIV replication. Our analysis reveals an enrichment of anti-inflammatory drugs in drug combinations that synergize against HIV. As inflammation accompanies HIV infection, these findings indicate that inhibiting inflammation could curb HIV propagation. Multiple drug pairs identified in this study, including various glucocorticoids and nitazoxanide (NTZ), synergize by targeting different steps in the HIV life cycle. MuSIC can be applied to a wide variety of disease-relevant screens to facilitate efficient identification of compound combinations. - Natture Biotech. 2012
shRNAs can trigger effective silencing of gene expression in mammalian cells, thereby providing powerful tools for genetic studies, as well as potential therapeutic strategies. Specific shRNAs can interfere with the replication of pathogenic viruses and are currently being tested as antiviral therapies in clinical trials. However, this effort is hindered by our inability to systematically and accurately identify potent shRNAs for viral genomes. Here we apply a recently developed highly parallel sensor assay to identify potent shRNAs for HIV, hepatitis C virus (HCV), and influenza. We observe known and previously unknown sequence features that dictate shRNAs efficiency. Validation using HIV and HCV cell culture models demonstrates very high potency of the top-scoring shRNAs. Comparing our data with the secondary structure of HIV shows that shRNA efficacy is strongly affected by the secondary structure at the target RNA site. Artificially introducing secondary structure to the target site markedly reduces shRNA silencing. In addition, we observe that HCV has distinct sequence features that bias HCV- targeting shRNAs toward lower efficacy. Our results facilitate further development of shRNA based antiviral therapies and improve our understanding and ability to predict efficient shRNAs. - PNAS 2012
Small interfering RNA (siRNA) are widely used to infer gene function. Here, insights in the equilibrium of siRNA-target hybridization are used for selection of efficient siRNA. The accessibilities of siRNA and target mRNA for hybridization, as measured by folding free energy change, are shown to be significantly correlated with efficacy. For this study, a partition function calculation that considers all possible secondary structures is used to predict target site accessibility; a significant improvement over calculations that consider only the predicted lowest free energy structure or a set of low free energy structures. The predicted thermodynamic features, in addition to siRNA sequence features, are used as input for a support vector machine that selects functional siRNA. The method works well for predicting efficient siRNA (efficacy 70%) in a large siRNA data set from Novartis. The positive predic- tive value (percentage of sites predicted to be efficient for silencing that are) is as high as 87.6%. The sensitivity and specificity are 22.7 and 96.5%, respectively. When tested on data from different sources, the positive predictive value increased 8.1% by adding equilibrium terms to 25 local sequence features. - Nucleic Acids Res. 2008a
相关专利
技术成果相关报道
联系方式
| 地址: | 清华大学 生命科学学院,生物信息学“教育部重点实验室”, 北京,100084 |
|---|---|
| 办公电话: | +86-10-62789217 |
| E-mail: | lulab1@tsinghua.edu.cn | bio.lulab@gmail.com |
| 实验室主页: | http://lulab.life.tsinghua.edu.cn | http://www.ncRNAlab.org |